







Comment la médecine fondée sur les preuves s’est imposée
Publié en ligne le 23 juin 2025 - Histoire des sciences -
Au fil des escroqueries et scandales sanitaires survenus depuis le XIXe siècle, la question de la preuve en médecine est progressivement devenue centrale. Si la médecine fondée sur les preuves semble s’être imposée depuis quelques décennies, les récentes polémiques survenues durant la pandémie de Covid-19 nous rappellent que ce gold standard, cette méthode de référence, ne va pas encore toujours de soi…
Comment évaluer l’efficacité d’un traitement médical ? En 1865, le pionnier de la médecine moderne, le médecin, physiologiste et épistémologue français Claude Bernard, se posait déjà la question en ces termes : « Comment savoir si c’est le remède ou la nature qui a guéri ? » [1].

Depuis cette époque, les normes de preuve pour évaluer l’efficacité et la sécurité des traitements médicaux ont considérablement évolué. Une rupture s’est imposée dans les années 1990 avec l’avènement de la « médecine fondée sur les preuves » (en anglais evidence-based medicine) [2], qui a notamment permis un contrôle plus strict des médicaments [3]. Mais de quoi parle-t-on exactement ?
L’émergence d’une législation régulant les médicaments
Les États-Unis furent des pionniers dans le développement d’une régulation des médicaments [3]. C’est pourquoi ils serviront de référence dans cet article.
Jusqu’au début du XXe siècle, il n’existait aucun contrôle sur la production et la vente de médicaments. En 1886, on trouvait donc en vente libre des remèdes comme le Microbe Killer de William Radam, censé guérir toutes les maladies [4, 5]. Afin de promouvoir son produit, Radam usa d’un marketing novateur, faisant publier des publicités dans les journaux et rédigeant plusieurs livres. Exploitant l’intérêt pour les microbes suite à la découverte de l’agent de la tuberculose par Robert Koch en 1882, il exposa dans Microbes and the Microbe Killer [6] sa théorie d’une origine unique à toutes les maladies ainsi que les témoignages enthousiastes de ceux qui avaient utilisé son produit. Le Microbe Killer connut un grand succès et fut vendu dans le monde entier [5].
En 1902, une analyse réalisée par le Bureau of Chemistry du département de l’Agriculture dirigé par le chimiste Harvey Washington Wiley [7] révéla que le Microbe Killer n’était composé que d’acide sulfurique et de sulfate de zinc fortement dilués dans de l’eau.
Les études réalisées par l’équipe de Wiley, surnommée la « brigade des poisons » [8], démontrèrent également la toxicité de certains conservateurs alimentaires utilisés couramment à l’époque, comme l’acide borique, les borates (sels ou esters de l’acide borique) et une forme diluée de formaldéhyde appelée formol, ainsi que la toxicité de remèdes courants, comme le calomel et le bichlorure de mercure [9, 10], deux remèdes employés contre la syphilis qui contenaient du mercure, un métal lourd aux effets particulièrement délétères sur la santé. Ces résultats soulignèrent l’urgence d’une loi fédérale sur les aliments et les médicaments.
Par ailleurs, en 1906, la parution du roman The Jungle du journaliste Upton Sinclair, qui dénonçait les pratiques insalubres de l’industrie alimentaire [11], suscita une réaction du public en faveur d’une réglementation gouvernementale [12]. C’est cette même année 1906 qui vit l’adoption par le Congrès américain, après plus de 200 rejets en 27 ans, du texte de loi « Pure Food and Drugs Act » qui interdisait enfin l’étiquetage mensonger des aliments et des médicaments [13, 14]. Elle entraîna la création de la Food and Drug Administration (FDA), qui prit la suite du Bureau of Chemistry et dont les missions sont de s’assurer de la sécurité des aliments et des médicaments, de leur étiquetage honnête et précis, ainsi que de poursuivre la fraude.
En 1937, l’Elixir sulfanilamide, un antibiotique sulfamidé, causa un empoisonnement de masse et la mort de plus de cent personnes aux ÉtatsUnis [15]. Le scandale entraîna en 1938 l’adoption du « Federal Food, Drug, and Cosmetic Act » qui introduisit l’obligation faite aux fabricants de médicaments de démontrer que le médicament est sûr pour l’utilisation humaine [16].

Cependant, cette loi ne permettait pas encore de faire retirer du marché les nombreux médicaments intrinsèquement dangereux qui y étaient déjà présents et sa capacité à empêcher l’introduction de nouvelles préparations problématiques était limitée [17, 18]. En effet, la FDA ne disposait que de 60 jours pour faire la preuve de la dangerosité d’un nouveau médicament et empêcher sa commercialisation. En outre, elle ne disposait pas du pouvoir de faire respecter les bonnes pratiques de fabrication.
Il a fallu que survienne une nouvelle tragédie, celle de la thalidomide, pour que la législation soit renforcée. Vendu dans les années 1950 comme sédatif et anti-nauséeux destiné aux femmes enceintes, ce médicament fut associé à de graves malformations congénitales chez des milliers de nouveau-nés en Europe [19]. Ce drame poussa le Congrès américain à adopter en 1962 l’amendement Kefauver-Harris [18]. Celui-ci exigeait que tout médicament soit évalué avant sa mise sur le marché et fasse la preuve de son efficacité et de sa sécurité au moyen d’études cliniques rigoureuses.
En Europe, avant la création de l’Union européenne (UE), chaque pays disposait de sa propre législation. La directive 65/65/CEE du 26 janvier 1965 introduisit des exigences uniformes pour l’enregistrement des médicaments [20]. Mais ce n’est qu’en 1995 que fut créée l’Agence européenne du médicament (EMA), dont la mission principale est d’autoriser et de contrôler les médicaments dans l’UE [21].
La médecine fondée sur les preuves
En 1972, le médecin et épidémiologiste britannique Archibald Leman Cochrane [22] publia un important rapport critique sur les pratiques médicales [23]. Il y démontrait les faiblesses des décisions médicales basées sur l’expérience personnelle du médecin, toujours limitée et subjective, et recommandait de réaliser des essais cliniques contrôlés afin d’évaluer plus objectivement chaque traitement.
Dans un essai contrôlé, l’effet d’un médicament est comparé à celui d’un placebo, une substance « inerte » qui ressemble au médicament mais ne contient aucun ingrédient actif. Un médicament n’est estimé efficace que si ses effets positifs pour le patient sont significativement supérieurs à ceux du placebo. Dans le cas où un traitement existe déjà, un essai contrôlé peut également comparer le nouveau médicament à celui-ci.
De nombreux raffinements émergèrent pour augmenter la fiabilité des essais. Dans les essais « randomisés et en double insu » (ou « double aveugle », traduction de l’anglais double-blind), les participants recevant l’un ou l’autre des traitements sont choisis de manière aléatoire, et ni eux ni les chercheurs ne savent quel traitement est administré.
Cette pratique réduit le risque que les attentes des participants ou les préjugés des chercheurs n’influencent les résultats de l’étude, en raison respectivement de l’effet placebo et du biais de confirmation (tendance à accorder davantage de poids aux informations qui confirment nos croyances qu’à celles qui les infirment).
En 1980, la FDA exigea que la preuve de l’efficacité d’un médicament soit obtenue à partir d’essais randomisés en double aveugle contre placebo [24]. Ceux-ci devinrent le gold standard, le standard le plus élevé de l’évaluation des traitements médicaux.

Soulignons qu’il n’est toutefois pas toujours possible d’évaluer un traitement médical via un essai clinique, en particulier si celui-ci présente des risques et concerne des femmes enceintes ou des nouveau-nés. Pour pallier ce problème, des études de cohortes peuvent être réalisées. Les participants sont regroupés en fonction de leur exposition à un traitement ou à un facteur de risque, comme la consommation de tabac, et sont observés sur une période donnée. La comparaison entre les groupes exposés et non exposés permet de mettre en évidence les conséquences pour la santé d’une exposition spécifique.
Il fallut cependant attendre les années 1990, avec l’informatisation de la recherche médicale et le développement des banques de données accessibles via Internet, pour que commence à s’imposer la pratique d’une médecine fondée sur les preuves [2]. Celle-ci est définie par l’un de ses pionniers, le médecin et épidémiologiste américano-canadien David L. Sackett [25], comme « l’utilisation consciencieuse, explicite et judicieuse des meilleures preuves actuelles dans la prise de décisions concernant les soins aux patients » [26].
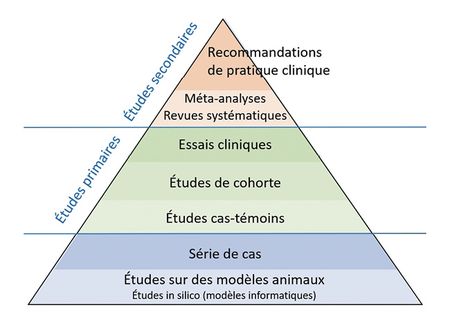
Le consensus médical se fonde sur une hiérarchie de niveaux de preuve.
Les recommandations de pratique clinique se fondent sur une analyse de la littérature scientifique (méta-analyses, revues systématiques). Ce sont les « études secondaires ».
Cette littérature scientifique est composée de comptes rendus d’études (études primaires). Ces dernières peuvent être de différentes natures :
- essais cliniques : études expérimentales où des participants sont assignés à des groupes pour tester l’efficacité et la sécurité d’un traitement ou d’une intervention ;
- études de cohorte : études observationnelles suivant un groupe de personnes sur une période pour examiner l’association entre une exposition et un événement de santé ;
- études cas-témoins : études rétrospectives comparant des individus malades (cas) à des individus sains (témoins) pour identifier des facteurs de risque potentiels.
Les essais cliniques (études d’intervention) ne sont pas toujours facilement réalisables (femmes enceintes, nourrissons, etc.). Les études de cohorte (études d’observation) peuvent apporter les preuves voulues, et sont parfois d’un très fort niveau [1]. C’est une étude de cohorte qui a démontré dans les années 1950 les effets nocifs du tabac. La pharmacovigilance repose largement sur des études d’observation et certains produits ont été ainsi démontrés comme dangereux (Mediator, Dépakine, etc.).
Référence
1 | Haut Conseil de la santé publique, « Les cohortes au niveau international : histoire et perspective », Actualité et dossier en santé publique n° 78, 2012.
Une pyramide des niveaux de preuves
La construction du consensus médical fondée sur les preuves repose sur deux grands principes : l’analyse de la totalité des études disponibles dans les banques de données et la hiérarchisation de celles-ci.
Cette hiérarchisation est souvent présentée sous la forme d’une pyramide des preuves où les études de suivi de cohortes de plusieurs milliers d’individus au cours du temps et les essais cliniques contrôlés et randomisés ont plus de valeur que les avis d’experts et les simples analyses de cas cliniques [27].
Les méta-analyses, analyses statistiques qui combinent les résultats de plusieurs essais cliniques, complètent le tableau. Elles sont souvent considérées comme la source de preuves la plus fiable, mais elles sont toutefois sujettes à des limitations [28]. En effet, il est rare que l’on dispose de toutes les données observationnelles et expérimentales nécessaires à l’évaluation rigoureuse d’un traitement et l’évaluation de ces données est inévitablement entachée d’une certaine subjectivité.

Le système GRADE (pour Grading of Recommendations, Assessment, Development, and Evaluations) est l’outil le plus largement adopté pour évaluer la qualité des données disponibles et émettre des recommandations concernant un traitement médical [29].
Dans ce système les données disponibles concernant un traitement sont classées en fonction de leur niveau de certitude. On distingue quatre niveaux. « Très faible » : l’effet réel est probablement nettement différent de l’effet estimé. « Bas » : l’effet réel peut être nettement différent de l’effet estimé. « Modéré » : les auteurs estiment que l’effet réel est probablement proche de l’effet estimé. « Haut » : les auteurs sont très confiants dans le fait que l’effet réel est similaire à l’effet estimé.
Ce classement fait appel à un ensemble d’outils permettant de mesurer les limitations et les biais des études disponibles. Par exemple, ces outils évaluent la cohérence des données, c’est-à-dire la concordance entre les différentes études disponibles. Ils prennent aussi en compte la puissance statistique des études publiées, c’est-à-dire le risque que certains résultats soient dus au hasard. Ils évaluent également les possibles « biais de publication », c’est-à-dire le fait que certaines études peuvent ne pas avoir été publiées parce qu’elles ne démontraient pas l’efficacité d’un traitement. La non-publication de ces « résultats négatifs » peut fortement biaiser les résultats des méta-analyses.
Le système GRADE aboutit à des recommandations qui peuvent être faibles ou fortes. Des recommandations fortes indiquent que presque toutes les personnes opteraient pour cette intervention. À l’inverse, des recommandations faibles suggèrent une probabilité élevée de variations dans les choix des personnes informées.
L’organisation Cochrane
La nécessité d’organiser et d’évaluer l’ensemble des informations concernant la recherche médicale et de les communiquer aux médecins a mené à l’émergence de l’organisation Cochrane (nommée initialement « Collaboration Cochrane » et aujourd’hui simplement « Cochrane »). Il s’agit d’une organisation internationale indépendante à but non lucratif qui regroupe plusieurs dizaines de milliers de volontaires (chercheurs, médecins, patients, professionnels de santé) dans plus de 190 pays et dispose d’un siège à l’Organisation mondiale de la santé [30].
Cochrane évalue les essais cliniques et les méta-analyses disponibles afin de produire des guides de pratique clinique qu’elle communique ensuite aux médecins. Ses travaux sont publiés dans la bibliothèque Cochrane et sont régulièrement actualisés [31].
Le consensus médical actuel est encore appelé à beaucoup évoluer. En effet, si l’on se réfère aux normes actuelles de la médecine fondée sur les preuves, la plupart des interventions médicales (94 %) ne sont pas étayées par des preuves de haute qualité [32]. Par intervention médicale, on entend toutes les décisions médicales. Les médicaments bien entendu, mais également les tests diagnostiques, les prises en charge psychiatriques, les interventions chirurgicales, etc. Le fait qu’elles ne soient pas encore suffisamment fondées sur les preuves n’impliquent évidemment pas qu’elles soient inefficaces ou qu’aucune preuve ne légitime leur usage.
Un changement de paradigme encore mal accepté
Les principes de la médecine fondée sur les preuves ont fait de la médecine une science. Ils ont permis de rompre avec la « médecine d’art » pratiquée depuis l’Antiquité, qui se caractérisait par un choix du traitement en grande partie basé sur l’expérience personnelle et l’intuition du médecin ainsi que ses a priori.
Toutefois, ce changement n’est pas encore unanimement accepté. Ainsi, durant la pandémie de Covid-19, on a pu assister à de violentes controverses médiatiques concernant l’hydroxychloroquine et les vaccins. La médecine fondée sur les preuves a été accusée par certains d’être au service de l’industrie pharmaceutique et contraire à l’éthique médicale (cette thèse fut notamment défendue par le microbiologiste Didier Raoult [33] et le sociologue Laurent Mucchielli [34]).
Cependant, comme nous l’avons vu, le développement de la médecine fondée sur les preuves n’est pas la conséquence des pressions de l’industrie. Au contraire, les lobbyistes des entreprises privés se sont souvent opposés à la mise en place d’une législation contraignante sur les médicaments, car la réalisation des essais cliniques retarde leur mise sur le marché et représente un coût très important pour les entreprises privées. En effet, le coût moyen pour valider un traitement est estimé à 33 millions de dollars [35], et plus de 90 % des traitements médicaux candidats échouent à traverser la « vallée de la mort » des essais cliniques [36], dont l’objectif est de démontrer leur innocuité et leur efficacité. Ainsi, chaque traitement qui n’est pas validé par les essais cliniques représente une perte financière très importante pour l’industrie.
Certes, la médecine fondée sur les preuves n’est pas parfaite : elle reste faillible et suscite toujours des critiques [37], notamment en raison des risques de conflits d’intérêts dus au financement des essais cliniques par l’industrie [38].
Pour améliorer sa fiabilité, diverses pistes ont été proposées. Il serait par exemple utile de valoriser la publication des résultats négatifs dans les journaux scientifiques afin de réduire les biais de publication dans les méta-analyses. Une autre piste serait de renforcer l’indépendance des universités et des chercheurs via un meilleur financement, ce qui réduirait les conflits d’intérêts entre les chercheurs et l’industrie.
En attendant, dans sa forme actuelle, la médecine fondée sur les preuves est ce dont nous disposons de plus efficace pour évaluer l’efficacité d’un traitement médical, lutter contre le charlatanisme et réduire le risque de catastrophes sanitaires.
1 | Bernard C, Introduction à l’étude de la médecine expérimentale, 1865.
2 | Ratnani I et al., “Evidence-based medicine : history, review, criticisms, and pitfalls”, Cureus, 2023, 15 :e35266.
3 | Meadows M, “Promoting safe & effective drugs for 100 years”, FDA Consumer magazine, février 2006.
4 | Museum of Health Care, « William Radam’s microbe killer », description, 2024. Sur mhc.andornot.net
5 | “Radam’s Microbe Killer : advertising cures for tuberculosis”, Circulating Now, 9 octobre 2015.
6 | Radam W, Microbes and the microbe killer, 1890.
7 | US Food and Drug Administration, “Harvey Washington Wiley”, biographie, 24 février 2020
8 | Bazaco MC, “The poison squad : one chemist’s single-minded crusade for food safety at the turn of the twentieth century”, Emerging Infectious Diseases, 2020, 26 :1639.
9 | “Calomel in Syphilis”, NEJM, 1831, 4 :112-4.
10 | Bland J, “The bichloride of mercury in syphilis and gonorrhœa”, The Lancet, 1841, 37 :408.
11 | Sinclair U, The Jungle, Doubleday, 1906.
12 | Klein C, “How Upton Sinclair’s ‘The Jungle’ led to us food safety reforms”, History, 10 mai 2023.
13 | Barkan ID, “Industry invites regulation : the passage of the Pure Food and Drug Act of 1906”, American Journal of Public Health, 1985, 75 :18-26.
14 | US Food and Drug History, “Part I : the 1906 Food and Drugs Act and Its Enforcement”, 2019.
15 | Ballentine C, “Sulfanilamide disaster”, FDA Consumer magazine, 1981.
16 | “Federal food, drug, and cosmetic act”, Public Law 118-83, 26 septembre 2024. Sur govinfo.gov
17 | US Food and Drug History, “80 Years of the Federal Food, Drug, and Cosmetic Act”, 2018.
18 | US Food and Drug Administration, “Kefauver-Harris amendments revolutionized drug development”, Consumer Updates, octobre 2012.
19 | Lenz W, “A short history of thalidomide embryopathy”, Teratology, 1988, 38 :203-15.
20 | Directive 65/65/CEE du Conseil du 26 janvier 1965 concernant le rapprochement des dispositions législatives, réglementaires et administratives, relatives aux spécialités pharmaceutiques, abrogée par directive 2001/83/CE du Parlement européen et du Conseil du 6 novembre 2001 instituant un code communautaire relatif aux médicaments à usage humain, 28 novembre 2001. Sur eu-lex.europa.eu
21 | Union européenne, « Agence européenne des médicaments (EMA) », présentation. Sur european-union.europa.eu
22 | Stavrou A et al.,“Archibald Cochrane (1909–1988) : the father of evidence-based medicine”, Interactive CardioVascular and Thoracic Surgery, 2014, 18 :121-4.
23 | Greenhalgh T, “Effectiveness and efficiency : random reflections on health services”, BMJ, 2004, 328 :529.
24 | Wampold BE et al., “The story of placebo effects in medicine : evidence in context”, Journal of Clinical Psychology, 2007, 63 :379-90.
25 | Collier R, “Dr. David Sackett, a giant among giants (19342015)”, CMAJ, 2015, 187 :640-1.
26 | Sackett DL, “Evidence-based medicine”, Seminars in Perinatology, 1997, 21 :3-5.
27 | Djulbegovic B, Guyatt GH, “Progress in evidence-based medicine : a quarter century on”, The Lancet, 2017, 390 :415-23.
28 | Berlin JA, Golub RM, “Meta-analysis as evidence : building a better pyramid”, Journal of the American Medical Association, 2014, 312 :603-6.
29 | BMJ, “What is GRADE ?”, page web.
30 | Le site de l’organisation Cochrane : consulté le 2 décembre 2024. Sur cochrane.org
31 | Le site de la bibliothèque Cochrane, consulté le 2 décembre 2024. Sur cochranelibrary.com
32 | Jeremy H et al., “Most healthcare interventions tested in Cochrane Reviews are not effective according to high quality evidence : a systematic review and meta-analysis”, Journal of Clinical Epidemiology, 2022, 148 :160-9.
33 | Raoult D, « Le médecin peut et doit réfléchir comme un médecin, et non pas comme un méthodologiste », tribune, Le Monde, 26 mars 2020.
34 | Mucchielli L, « Trafic d’influence : le rôle de l’industrie pharmaceutique dans la controverse sur le traitement médical de la Covid », Les Cahiers du CEDIMES, 2021, 16 :76-86.
35 | Martin L et al., “How much do clinical trials cost ?”, Nature Reviews Drug Discovery, 2017, 16 :381-2.
36 | Duxin S et al., “Why 90 % of clinical drug development fails and how to improve it ?”, Acta Pharmaceutica Sinica B, 2022,12 :3049-62.
37 | Djulbegovic B et al., “Epistemologic inquiries in evidencebased medicine”,Cancer Control, 2009, 16 :158-68.
38 | Jureidini J, McHenry LB, “The illusion of evidence based medicine”, BMJ, 2022, 376 :o702.
Publié dans le n° 351 de la revue

Partager cet article




Les auteurs
Éric Muraille

Biologiste et immunologiste. Il est directeur de recherche au FNRS, Université libre de Bruxelles (ULB).
Plus d'informationsElie Cogan

Professeur émérite de médecine interne, membre de l’Académie royale de médecine de Belgique, Université libre de (…)
Plus d'informationsHistoire des sciences

Une autre histoire de la lèpre dans les Amériques
Le 19 juin 2026
Comment la médecine fondée sur les preuves s’est imposée
Le 23 juin 2025
![[Conférence en ligne - Mardi 25 février 2025 à 20h00] Les transfusions sanguines : quand les prescrire ? Que craindre ?](local/cache-gd2/77/cc065f89480403b77b0f80a6a4178a.png?1740122862)











